Sickle Cell Disease (SCD)
What is Sickle Cell Disease (SCD)?
Sickle Cell Disease is an inherited form of anemia — a condition in which there is enough not healthy red blood cells to carry adequate oxygen throughout the body. It affects hemoglobin, the molecule in red blood cells that delivers oxygen to cells throughout the body. People with this disorder have an atypical hemoglobin molecule called hemoglobin S which makes the red blood cells get distorted under certain conditions into a sickle cell shape or crescent shape. This also makes it sticky, rigid and prone to getting trapped in small blood vessels which blocks blood from reaching different parts of the body. This can cause pain and tissue damage. The painful crises called vaso-occlusive crises (VOC) is the hallmark of the disease and leads to poor quality of life and eventually complications due to organ damage. These patients are also at risk for serious infections by encapsulated bacteria.
According to WHO estimates, Sickle Cell Disease is common among people in sub-Saharan Africa, India, Saudi Arabia and Mediterranean countries. The SCD trait is so widespread in sub-Saharan Africa, central India and eastern Saudi Arabia, that it has affected up to 40% of these populations.
SCD is most prevalent in Africa as the SCD gene evolved to provide a measure of protection against falciparum malaria which is endemic to the region. However, though a single gene may provide protection, inheriting two copies of the mutated gene from both parents result in the deadly form of SCD which is incompatible with life.
Patterns of Inheritance
SCD is caused by a point mutation in the β-globin chain of hemoglobin causing glutamic acid to be replaced with the amino acid valine at the sixth position to form hemoglobin S (HbS). This is called a structural hemoglobin variant. Some patients may present with compound heterzygote states due to co-inheritance of Hb S from one parent and Beta Thalassemia or other thalassemias or variant hemoglobins from the other parent.
- Hemoglobin Sβ0 thalassemia
- Hemoglobin Sβ+ thalassemia
- Hemoglobin SC
- Hemoglobin SD
- Hemoglobin SE
- Hemoglobin SS
The hemoglobin gene is found on the short arm of chromosome 11. Under conditions of dehydration, fever or low-oxygen conditions (being at high altitude, for example), the absence of a polar amino acid at position six of the β-globin chain promotes the non-covalent polymerisation (aggregation) of hemoglobin which distorts red blood cells into a sickle shape and decreases their elasticity.
SCD is inherited in an autosomal recessive pattern which means that both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. In SCD, a person who carries one copy of the mutated gene is said to be a carrier for the condition.
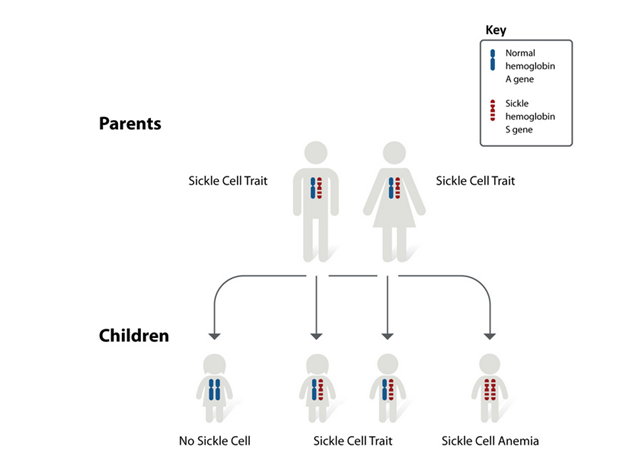
Image source: Pixabay
Symptoms and Diagnosis
Symptoms
Symptoms of SCD usually show up at a young age. They may appear in babies as early as 4 months old, but generally occur around the 6-month mark.
- Fussiness in babies
- Fatigue and tiredness
- Jaundice - yellowing of the eyes and skin
- Swelling and pain in hands and feet
- Frequent infections- pneumonia
- Backpain or bone pains
- Pallor or paleness
- Crises- acute chest syndrome
- Vaso-occlusive crisis etc
Children are only at risk for Sickle Cell Disease if both parents carry sickle cell trait. A blood test called hemoglobin electrophoresis can also determine which type you might carry.
Image source: Pixabay
Diagnosis
In children and adults, one or more of the following procedures may also be used to diagnose Sickle Cell Disease.
Detailed patient history
This condition often first appears as acute pain in the hands and feet. Patients may also have
- Severe pain in the hands, bones, back
- Anemia
- Painful enlargement of the spleen
- Growth problems
- Respiratory and other infections
- Ulcers of the legs
- Heart problems
- Stroke and transient ischemic episodes
- Lung problems
- Kidney problems
Your doctor may want to test you for SCD if you have any of the symptoms mentioned above.
Blood Tests
Several blood tests can be used to look for SCD:
- Blood counts can reveal an abnormal Hb level in the range of 6 to 8 grams per decilitre
- Blood films may show RBCs that appear as irregularly contracted cells
- Sickle solubility tests look for the presence of Hb S
- HPLC/Hb capillary zone electrophoresis or Hb electrophoresis can confirm the diagnosis of Sickle Cell Disease. These methods measure the amount of hemoglobin S in the blood
- Molecular testing to identify the SCD genotype
Prenatal testing is available to look for the sickle cell gene in the mother’s amniotic fluid.
By regular health maintenance and parental counseling, the early high mortality rates seen in these children has gone down. Physicians need to be aware of fever, jaundice, pallor and should monitor the spleen size at each visit. Basic tests like complete blood count, reticulocyte count, routine biochemistry tests like LFT, RFT are useful to monitor the patients. An estimation of HbF is necessary. Other tests such as Transcranial Doppler ultrasonography (TCD), magnetic resonance imaging (MRI) with or without angiography and neuro-psychometric (NPM) studies may be done if provision is available or the child can be referred to a centre where it can be done.
Educational material should be given to the caregiver and older children, so they understand about the disease and especially about fever in SCD. Sickle cell carriers usually have mild disease, but may need follow-ups for regular health maintenance. Some will need intervention for fever and pain. Genetic counseling should be made available to all carriers.

Image source: Pixabay
Current Management
A number of different treatments are available for SCD:
- Hydroxyurea helps to increase production of fetal hemoglobin. This can reduce vaso-occlusive crises and may reduce the number of blood transfusions needed by increasing the hemoglobin F. Many trials have shown the effectiveness of this medication. It is usually started at a lower dose of 10mg/ kg of body weight and usually it increases hemoglobin and alleviates VOC crises. If responses are not satisfactory, the dose can be increased the child/adult responds well. The medicine requires regular monitoring and should be taken under the supervision of a doctor.
- Immunizations can help prevent infections. Patients tend to have lower immunity in SCD
- Staying well hydrated. The red blood cells are more likely to deform and assume the sickle shape if the individual is dehydrated
- Pneumococcal immunization and penicillin prophylaxis. Other vaccines like H. influenza and meningococcal vaccine are also given. Routine childhood vaccines are necessary
- Treating underlying or associated infections is important as the stress of an infection can result in a sickle cell crisis. An infection may also result as a complication of a crisis
- Blood transfusions improve transport of oxygen and nutrients as needed. Packed red cells are removed from donated blood and given to patients. However, monitoring and care is needed as patient may develop allo-antibodies to the minor red blood cell antigens
- Supplemental oxygen is given through a mask. It makes breathing easier and improves oxygen levels in the blood
- Pain medication is used to relieve pain during a sickle crisis. You may need over-the-counter drugs or stronger prescription pain medication
It is important to understand when patients need to be admitted for VOC. Patients can develop acute chest syndrome, pneumonias, aplastic crises after parvovirus B19 infections, allo-immunization due to blood transfusions, sepsis due to functional asplenia, iron overload and even splenic sequestration crises.
Bone marrow transplant has been used to treat SCD. Children younger than 16 years of age who have severe complications and have a matching donor are the best candidates. Indications for allogeneic Hematopoietic Stem Cell Transplant (HSCT) for Sickle Cell Disease as suggested by Walters et al (3):
- Stroke or central nervous system event lasting longer than 24 hours, acute chest syndrome with recurrent hospitalizations or previous exchange transfusions
- Recurrent vaso-occlusive pain (more than 2 episodes per year over several years) or recurrent priapism
- Impaired neuropsychological function with abnormal cerebral MRI scan
- Stage I or II sickle lung disease
- Sickle nephropathy (moderate or severe proteinuria or a glomerular filtration rate 30 to 50% of the predicted normal value)
- Bilateral proliferative retinopathy with major visual impairment in at least one eye
- Osteonecrosis of multiple joints
- Red-cell allo-immunization during long-term transfusion therapy
Investigational Therapies
Recently approved drugs such as L-glutamine have shown great improvement in managing inflammatory adhesion during painful crisis and managing red blood cell health.
Several newer drugs like crizanlizumab-tmca/voxelotor are approved for patients and may be useful in preventing VOC by reducing the pro-inflammatory state.
Currently gene therapy is also being studied as a permanent cure for SCD. Gene therapy involves replacing the defective gene with a normal gene through the help of load carrying viral vectors. The normal gene will be able to produce sufficient quantities of the hemoglobin and prevent the mutated form of hemoglobin from causing inflammatory events and painful crisis.
Gene editing techniques are also being studied and explored as a permanent cure for SCD. This involves reactivating the gene responsible for fetal hemoglobin production, BCLA11. After collecting blood stem cells from the patient, it is edited to reactivate the production of fetal hemoglobin and then infused back into the patient. Increasing the fetal hemoglobin levels boosts blood production and removes the need for transfusion dependency and prevents inflammatory and thromboembolic events.
References
Description, Types, Causes, Diagnosis, Treatment
https://rarediseases.info.nih.gov/diseases/8614/sickle-cell-anemia
https://ghr.nlm.nih.gov/condition/sickle-cell-disease#diagnosis
https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia
https://www.nhlbi.nih.gov/health-topics/sickle-cell-disease
Etiology
https://thalassemia.com/genetics-inheritance.aspx#gsc.tab=0
https://rarediseases.info.nih.gov/diseases/871/beta-thalassemia
https://www.ncbi.nlm.nih.gov/books/NBK22200/
Epidemiology
https://apps.who.int/gb/archive/pdf_files/WHA59/A59_9-en.pdf
http://www.scdcoalition.org/priorities/global.html
Gene Therapy
Standard of care guidelines
National Institutes of Health
https://www.nhlbi.nih.gov/files/docs/guidelines/sc_mngt.pdf
Centers for Disease Control and Preventio
https://www.cdc.gov/ncbddd/sicklecell/recommendations.html
ASH Clinical Practice Guidelines on Sickle Cell Disease
https://www.hematology.org/education/clinicians/guidelines-and-quality-care/clinical-practice-guidelines/sickle-cell-disease-guidelines
Sickle Cell Society
https://www.sicklecellsociety.org/wp-content/uploads/2018/05/Standards-for-the-Clinical-Care-of-Adults-with-Sickle-Cell-in-the-UK-2018.pdf
American Society of Hematology 2020
https://ashpublications.org/bloodadvances/article/4/8/1554/454384/American-Society-of-Hematology-2020-guidelines-for
National Resources
National Alliance of Sickle Cell Organizations
Address:
Piplafata, Besa road,
Nagpur 441 614, Maharasthra
Contact : Mr Gautam Dongre
Phone: +91 94217 09245 or +91 77218 19207
Website - www.indiascdalliance.org
Disease Videos
Sickle Cell Disease - causes, symptoms, diagnosis, treatment & pathology
What Is Sickle Cell Disease and How Do You Get It?
What is Sickle Cell Disease
Keywords
Sickle Cell Disease, Hemoglobin, Carriers, Minors, Trait, Point Mutation, HBS Gene, Bone Marrow Transplant (BMT), Blood Transfusion, Hydroxyurea, L-glutamine, Gene Therapy, Gene Editing, Foetal Hemoglobin, BCLA11 Gene